Tools
Preprocessing
qc_filter
Tools for quality control.
- sctoolbox.tools.qc_filter.calculate_qc_metrics(adata: AnnData, percent_top: list[int] | None = None, inplace: bool = False, **kwargs: Any) AnnData | None[source]
Calculate the qc metrics using scanpy.pp.calculate_qc_metrics.
- Parameters:
adata (sc.AnnData) – Anndata object the quality metrics are added to.
percent_top (Optional[list[int]], default None) – Which proportions of top genes to cover.
inplace (bool, default False) – If the anndata object should be modified in place.
**kwargs (Any) – Additional parameters forwarded to scanpy.pp.calculate_qc_metrics.
- Returns:
Returns anndata object with added quality metrics to .obs and .var. Returns None if inplace=True.
- Return type:
Optional[sc.AnnData]
See also
scanpy.pp.calculate_qc_metricshttps//scanpy.readthedocs.io/en/stable/generated/scanpy.pp.calculate_qc_metrics.html
Examples
import scanpy as sc import sctoolbox as sct adata = sc.datasets.pbmc3k() print("Columns in .obs before 'calculate_qc_metrics':", adata.obs.columns.tolist()) sct.tools.calculate_qc_metrics(adata, inplace=True) print("Columns in .obs after 'calculate_qc_metrics':", adata.obs.columns.tolist())
Columns in .obs before 'calculate_qc_metrics': [] Columns in .obs after 'calculate_qc_metrics': ['n_genes', 'log1p_n_genes', 'total_counts', 'log1p_total_counts']
- sctoolbox.tools.qc_filter.predict_cell_cycle(adata: AnnData, species: str | None, s_genes: str | list[str] | None = None, g2m_genes: str | list[str] | None = None, inplace: bool = True) AnnData | None[source]
Assign a score and a phase to each cell depending on the expression of cell cycle genes.
- Parameters:
adata (sc.AnnData) – Anndata object to predict cell cycle on.
species (Optional[str]) – The species of data. Available species are: human, mouse, rat and zebrafish. If both s_genes and g2m_genes are given, set species=None, otherwise species is ignored.
s_genes (Optional[str | list[str]], default None) – If no species is given or desired species is not supported, you can provide a list of genes for the S-phase or a txt file containing one gene in each row. If only s_genes is provided and species is a supported input, the default g2m_genes list will be used, otherwise the function will not run.
g2m_genes (Optional[str | list[str]], default None) – If no species is given or desired species is not supported, you can provide a list of genes for the G2M-phase or a txt file containing one gene per row. If only g2m_genes is provided and species is a supported input, the default s_genes list will be used, otherwise the function will not run.
inplace (bool, default True) – if True, add new columns to the original anndata object.
- Returns:
If inplace is False, return a copy of anndata object with the new column in the obs table.
- Return type:
Optional[sc.AnnData]
- Raises:
ValueError: – 1: If s_genes or g2m_genes is not None and not of type list. 2: If no cellcycle genes available for the given species. 3. If given species is not supported and s_genes or g2m_genes are not given.
- sctoolbox.tools.qc_filter.estimate_doublets(adata: AnnData, threshold: float = 0.25, inplace: bool = True, plot: bool = True, groupby: str | None = None, threads: int = 4, fill_na: bool = True, **kwargs: Any) AnnData | None[source]
Estimate doublet cells using scrublet.
Adds additional columns “doublet_score” and “predicted_doublet” in adata.obs, as well as a “scrublet” key in adata.uns.
- Parameters:
adata (sc.AnnData) – Anndata object to estimate doublets for.
threshold (float, default 0.25) – Threshold for doublet detection.
inplace (bool, default True) – Whether to estimate doublets inplace or not.
plot (bool, default True) – Whether to plot the doublet score distribution.
groupby (Optional[str], default None) – Key in adata.obs to use for batching during doublet estimation. If threads > 1, the adata is split into separate runs across threads. Otherwise each batch is run separately.
threads (int, default 4) – Number of threads to use.
fill_na (bool, default True) – If True, replaces NA values returned by scrublet with 0 and False. Scrublet returns NA if it cannot calculate a doublet score. Keep in mind that this does not mean that it is no doublet. By setting this parameter true it is assmuned that it is no doublet.
**kwargs (Any) – Additional arguments are passed to scanpy.external.pp.scrublet.
Notes
Groupby should be set if the adata consists of multiple samples, as this improves the doublet estimation.
- Returns:
If inplace is False, the function returns a copy of the adata object. If inplace is True, the function returns None.
- Return type:
Optional[sc.AnnData]
- sctoolbox.tools.qc_filter.predict_sex(adata: AnnData, groupby: str, gene: str = 'Xist', gene_column: str | None = None, threshold: float = 0.3, plot: bool = True, save: str | None = None, **kwargs: Any) None[source]
Predict sex based on expression of Xist (or another gene).
- Parameters:
adata (sc.AnnData) – An anndata object to predict sex for.
groupby (str) – Column in adata.obs to group by.
gene (str, default "Xist") – Name of a female-specific gene to use for estimating Male/Female split.
gene_column (Optional[str], default None) – Name of the column in adata.var that contains the gene names. If not provided, adata.var.index is used.
threshold (float, default 0.3) – Threshold for the minimum fraction of cells expressing the gene for the group to be considered “Female”.
plot (bool, default True) – Whether to plot the distribution of gene expression per group.
save (Optional[str], default None) – If provided, the plot will be saved to this path.
**kwargs (Any) – Additional arguments are passed to scanpy.pl.violin.
Notes
adata.X will be converted to numpy.ndarray if it is of type numpy.matrix.
- Return type:
None
- sctoolbox.tools.qc_filter.automatic_thresholds(adata: AnnData, which: Literal['obs', 'var'] = 'obs', groupby: str | None = None, columns: list[str] | None = None) dict[str, dict[str, float | dict[str, float]]][source]
Get automatic thresholds for multiple data columns in adata.obs or adata.var.
- Parameters:
adata (sc.AnnData) – Anndata object to find thresholds for.
which (Literal["obs", "var"], default "obs") – Which data to find thresholds for. Either “obs” or “var”.
groupby (Optional[str], default None) – Group rows by the column given in ‘groupby’ to find thresholds independently per group
columns (Optional[list[str]], default None) – Columns to calculate automatic thresholds for. If None, will take all numeric columns.
- Returns:
A dict containing thresholds for each data column, either grouped by groupby or directly containing “min” and “max” per column.
- Return type:
dict[str, dict[str, Union[float, dict[str, float]]]]
- Raises:
ValueError: – If which is not set to ‘obs’ or ‘var’
- sctoolbox.tools.qc_filter.thresholds_as_table(threshold_dict: dict[str, dict[str, float | int | dict[str, int | float]]]) DataFrame[source]
Show the threshold dictionary as a table.
- Parameters:
threshold_dict (dict[str, dict[str, float | int | dict[str, int | float]]]) – Dictionary with thresholds.
- Return type:
pd.DataFrame
- sctoolbox.tools.qc_filter.validate_threshold_dict(table: DataFrame, thresholds: dict[str, dict[str, int | float] | dict[str, dict[str, int | float]]], groupby: str | None = None) None[source]
Validate threshold dictionary.
Thresholds can be in the format:
thresholds = {"chrM_percent": {"min": 0, "max": 10}, "total_reads": {"min": 1000}}
Or per group in ‘groupby’:
thresholds = {"chrM_percent": { "Sample1": {"min": 0, "max": 10}, "Sample2": {"max": 5} }, "total_reads": {"min": 1000}}
- Parameters:
table (pd.DataFrame) – Table to validate thresholds for.
thresholds (dict[str, dict[str, int | float] | dict[str, dict[str, int | float]]]) – Dictionary of thresholds to validate.
groupby (Optional[str], default None) – Column for grouping thresholds.
- Raises:
ValueError – If the threshold dict is not valid.
- sctoolbox.tools.qc_filter.get_thresholds_wrapper(adata: AnnData, manual_thresholds: dict, only_automatic_thresholds: bool = True, groupby: str | None = None) dict[str, dict[str, float | int | dict[str, float | int]]][source]
Get the thresholds for the filtering.
- Parameters:
adata (sc.AnnData) – Anndata object to find QC thresholds for.
manual_thresholds (dict[str, dict[str, Union[float, dict[str, float]]]]) – Dictionary containing manually set thresholds
only_automatic_thresholds (bool, default True) – If True, only set automatic thresholds.
groupby (Optional[str], default None) – Group cells by column in adata.obs.
- Returns:
Dictionary containing the thresholds
- Return type:
dict[str, dict[str, Union[float | int, dict[str, float | int]]]]
- sctoolbox.tools.qc_filter.get_keys(adata: AnnData, manual_thresholds: dict[str, Any]) dict[str, dict[str, float | int | dict[str, float | int]]][source]
Get threshold dictionary with keys that overlap with adata.obs.columns.
- Parameters:
adata (sc.AnnData) – Anndata object
manual_thresholds (dict[str, Any]) – Dictionary with adata.obs colums as keys.
- Returns:
Dictionary with key - adata.obs.column overlap
- Return type:
dict[str, dict[str, Union[float | int, dict[str, float | int]]]]
- sctoolbox.tools.qc_filter.get_mean_thresholds(thresholds: dict[str, Any]) dict[str, Any][source]
Convert grouped thresholds to global thresholds by taking the mean across groups.
- sctoolbox.tools.qc_filter.apply_qc_thresholds(adata: AnnData, thresholds: dict[str, Any], which: Literal['obs', 'var'] = 'obs', groupby: str | None = None, inplace: bool = True) AnnData | None[source]
Apply QC thresholds to anndata object.
- Parameters:
adata (sc.AnnData) – Anndata object to filter.
thresholds (dict[str, Any]) – Dictionary of thresholds to apply.
which (Literal["obs", "var"], default 'obs') – Which table to filter on. Must be one of “obs” / “var”.
groupby (Optional[str], default None) – Column in table to group by.
inplace (bool, default True) – Change adata inplace or return a changed copy.
- Returns:
Anndata object with QC thresholds applied.
- Return type:
Optional[sc.AnnData]
- Raises:
ValueError: – 1: If the keys in thresholds do not match with the columns in adata.[which]. 2: If grouped thesholds are not found. For example do not contain min and max values. 3: If thresholds do not contain min and max values.
- sctoolbox.tools.qc_filter.filter_cells(adata: AnnData, cells: str | list[str], remove_bool: bool = True, inplace: bool = True) AnnData | None[source]
Remove cells from anndata object.
- Parameters:
adata (sc.AnnData) – Anndata object to filter.
cells (str | list[str]) – A column in .obs containing boolean indicators or a list of cells to remove from object .obs table.
remove_bool (bool, default True) – Is used if genes is a column in .obs table. If True, remove cells that are True. If False, remove cells that are False.
inplace (bool, default True) – If True, filter inplace. If False, return filtered adata object.
- Returns:
If inplace is False, returns the filtered Anndata object. If inplace is True, returns None.
- Return type:
Optional[sc.AnnData]
- sctoolbox.tools.qc_filter.filter_genes(adata: AnnData, genes: str | list[str], remove_bool: bool = True, inplace: bool = True) AnnData | None[source]
Remove genes from adata object.
- Parameters:
adata (sc.AnnData) – Annotated data matrix object to filter
genes (str | list[str]) – A column containing boolean indicators or a list of genes to remove from object .var table.
remove_bool (bool, default True) – Is used if genes is a column in .var table. If True, remove genes that are True. If False, remove genes that are False.
inplace (bool, default True) – If True, filter inplace. If False, return filtered adata object.
- Returns:
If inplace is False, returns the filtered Anndata object. If inplace is True, returns None.
- Return type:
Optional[sc.AnnData]
highly_variable
Tools to calculate and annotate highly variable genes.
- sctoolbox.tools.highly_variable.annot_HVG(anndata: AnnData, min_mean: float = 0.0125, max_iterations: int = 10, hvg_range: tuple[int, int] = (1000, 5000), step: int | float = 10, inplace: bool = True, save: str | None = None, **kwargs: Any) AnnData | None[source]
Annotate highly variable genes (HVG). Tries to annotate in given range of HVGs, by gradually in-/ decreasing min_mean of scanpy.pp.highly_variable_genes.
- Parameters:
anndata (sc.AnnData) – Anndata object to annotate.
min_mean (float, default 0.0125) – Starting min_mean parameter for finding HVGs.
max_iterations (int, default 10) – Maximum number of min_mean adjustments.
hvg_range (Tuple[int, int], default (1000, 5000)) – Number of HVGs should be in the given range. Will issue a warning if result is not in range. Default limits are chosen as proposed by https://doi.org/10.15252/msb.20188746.
step (int | float, default 10) – Value min_mean is adjusted by in each iteration. Will divide min_value (below range) or multiply (above range) by this value.
inplace (bool, default False) – Whether the anndata object is modified inplace.
save (Optional[str], default None) – Path to save the plot to. If None, the plot is not saved.
**kwargs (Any) – Additional arguments forwarded to scanpy.pp.highly_variable_genes().
Notes
Logarithmized data is expected.
- Returns:
Adds annotation of HVG to anndata object. Information is added to Anndata.var[“highly_variable”]. If inplace is False, the function returns None Else returns a chagned copy of the input anndata object.
- Return type:
Optional[sc.AnnData]
- sctoolbox.tools.highly_variable.get_variable_features(adata: AnnData, max_cells: int | float | None = None, min_cells: int | float | None = 0, show: bool = True, inplace: bool = True) AnnData | None[source]
Get the highly variable features of anndata object. Adds the column “highly_variable” to adata.var. If show is True, the plot is shown.
- Parameters:
adata (sc.AnnData) – The anndata object containing counts for variables.
max_cells (Optional[float | int], default None) – The maximum variability score to set as threshold. Defaults to knee estimated threshold.
min_cells (Optional[float | int], default 0) – The minimum variability score to set as threshold.
show (bool, default True) – Show plot of variability scores and thresholds.
inplace (bool, default True) – If True, the anndata object is modified. Otherwise, a new anndata object is returned.
Notes
Designed for scATAC-seq data
- Raises:
KeyError – If adata.var[‘n_cells_by_counts’] is not available.
- Returns:
If inplace is False, the function returns None If inplace is True, the function returns an anndata object.
- Return type:
Optional[sc.AnnData]
dim_reduction
Tools for dimensionality reduction with PCA/SVD.
- sctoolbox.tools.dim_reduction.compute_PCA(anndata: AnnData, use_highly_variable: bool = True, inplace: bool = False, **kwargs: Any) AnnData | None[source]
Compute a principal component analysis.
- Parameters:
anndata (sc.AnnData) – Anndata object to add the PCA to.
use_highly_variable (bool, default True) – If true, use highly variable genes to compute PCA.
inplace (bool, default False) – Whether the anndata object is modified inplace.
**kwargs (Any) – Additional parameters forwarded to scanpy.pp.pca().
- Returns:
Returns anndata object with PCA components. Or None if inplace = True.
- Return type:
Optional[sc.AnnData]
- sctoolbox.tools.dim_reduction.lsi(data: AnnData, scale_embeddings: bool = True, n_comps: int = 50, use_highly_variable: bool = False) None[source]
Run Latent Semantic Indexing for dimensionality reduction.
Values represent the similarity of cells in the original space. doi: 10.3389/fphys.2013.00008
- Parameters:
data (sc.AnnData) – AnnData object with peak counts.
scale_embeddings (bool, default True) – Scale embeddings to zero mean and unit variance.
n_comps (int, default 50) – Number of components to calculate with SVD.
use_highly_variable (bool, default True) – If true, use highly variable genes to compute LSI.
- Raises:
ValueError: – If highly variable genes are used and not found in adata.var[‘highly_variable’].
Notes
Function is taken from muon package.
- sctoolbox.tools.dim_reduction.apply_svd(adata: AnnData, layer: str | None = None) AnnData[source]
Singular value decomposition of anndata object.
- Parameters:
adata (sc.AnnData) – The anndata object to be decomposed.
layer (Optional[str], default None) – The layer to be decomposed. If None, the layer is set to “X”.
- Returns:
The decomposed anndata object containing .obsm, .varm and .uns information.
- Return type:
sc.AnnData
- sctoolbox.tools.dim_reduction.define_PC(anndata: AnnData) int[source]
Define threshold for most variable PCA components.
Note: Function expects PCA to be computed beforehand.
- Parameters:
anndata (sc.AnnData) – Anndata object with PCA to get significant PCs threshold from.
- Returns:
An int representing the number of PCs until elbow, defining PCs with significant variance.
- Return type:
int
- Raises:
ValueError: – If PCA is not found in anndata.
Deprecated since version 0.5: This will be removed in 0.7. Use the ‘sctoolbox.tools.propose_pcs’ function instead.
- sctoolbox.tools.dim_reduction.propose_pcs(anndata: AnnData, how: list[Literal['variance', 'cumulative variance', 'correlation']] = ['variance', 'correlation'], var_method: Literal['knee', 'percent'] = 'percent', perc_thresh: int | float = 30, corr_thresh: float = 0.3, corr_kwargs: dict | None = {}) list[int][source]
Propose a selection of PCs that can be used for further analysis.
Note: Function expects PCA to be computed beforehand.
- Parameters:
anndata (sc.AnnData) – Anndata object with PCA to get PCs from.
how (List[Literal["variance", "cumulative variance", "correlation"]], default ["variance", "correlation"]) – Values to use for PC proposal. Will independently apply filters to all selected methods and return the union of PCs.
var_method (Literal["knee", "percent"], default "percent") – Either define a threshold based on a knee algorithm or use the percentile.
perc_thresh (Union[int, float], default 30) – Percentile threshold of the PCs that should be included. Only for var_method=”percent” and expects a value from 0-100.
corr_thresh (float, default 0.3) – Filter PCs with a correlation greater than the given value.
corr_kwargs (Optional(dict), default None) – Parameters forwarded to sctoolbox.tools.correlation_matrix.
- Returns:
List of PCs proposed for further use.
- Return type:
List[int]
- Raises:
ValueError: – If PCA is not found in anndata.
- sctoolbox.tools.dim_reduction.subset_PCA(adata: AnnData, n_pcs: int | None = None, start: int = 0, select: list[int] | None = None, inplace: bool = True) AnnData | None[source]
Subset the PCA coordinates in adata.obsm[“X_pca”] to the given number of pcs.
Additionally, subset the PCs in adata.varm[“PCs”] and the variance ratio in adata.uns[“pca”][“variance_ratio”].
- Parameters:
adata (sc.AnnData) – Anndata object containing the PCA coordinates.
n_pcs (Optional[int], default None) – Number of PCs to keep.
start (int, default 0) – Index (0-based) of the first PC to keep. E.g. if start = 1 and n_pcs = 10, you will exclude the first PC to keep 9 PCs.
select (Optional[List[int]], default None) – Provide a list of PC numbers to keep. E.g. [2, 3, 5] will select the second, third and fifth PC. Will overwrite the n_pcs and start parameter.
inplace (bool, default True) – Whether to work inplace on the anndata object.
- Returns:
Anndata object with the subsetted PCA coordinates. Or None if inplace = True.
- Return type:
Optional[sc.AnnData]
norm_correct
Normalization and correction tools.
- sctoolbox.tools.norm_correct.atac_norm(*args: Any, **kwargs: Any)[source]
Normalize ATAC data - deprecated functionality. Use normalize_adata instead.
- sctoolbox.tools.norm_correct.normalize_adata(adata: AnnData, method: str | list[str], exclude_highly_expressed: bool = True, use_highly_variable: bool = False, target_sum: int | None = None) dict[str, AnnData][source]
Normalize the count matrix and calculate dimension reduction using different methods.
- Parameters:
adata (sc.AnnData) – Annotated data matrix.
method (str | list[str]) – Normalization method. Either ‘total’ and/or ‘tfidf’. - ‘total’: Performs normalization for total counts, log1p and PCA. - ‘tfidf’: Performs TFIDF normalization and LSI (corresponds to PCA). This method is often used for scATAC-seq data.
exclude_highly_expressed (bool, default True) – Parameter for sc.pp.normalize_total. Decision to exclude highly expressed genes (HEG) from total normalization.
use_highly_variable (bool, default False) – Parameter for sc.pp.pca and lsi. Decision to use highly variable genes for PCA/LSI.
target_sum (Optional[int], default None) – Parameter for sc.pp.normalize_total. Decide the target sum of each cell after normalization.
- Returns:
Dictionary containing method name as key, and anndata as values. Each anndata is the annotated data matrix with normalized count matrix and PCA/LSI calculated.
- Return type:
dict[str, sc.AnnData]
- Raises:
ValueError – If method is not valid. Needs to be either ‘total’ or ‘tfidf’.
- sctoolbox.tools.norm_correct.tfidf(data: AnnData, log_tf: bool = True, log_idf: bool = True, log_tfidf: bool = False, scale_factor: int = 10000) None[source]
Transform peak counts with TF-IDF (Term Frequency - Inverse Document Frequency).
TF: peak counts are normalised by total number of counts per cell. DF: total number of counts for each peak. IDF: number of cells divided by DF. By default, log(TF) * log(IDF) is returned.
- Parameters:
data (sc.AnnData) – AnnData object with peak counts.
log_tf (bool, default True) – Log-transform TF term if True.
log_idf (bool, default True) – Log-transform IDF term if True.
log_tfidf (bool, default Frue) – Log-transform TF*IDF term if True. Can only be used when log_tf and log_idf are False.
scale_factor (int, default 1e4) – Scale factor to multiply the TF-IDF matrix by.
Notes
Function is from the muon package.
- Raises:
AttributeError: – log(TF*IDF) requires log(TF) and log(IDF) to be False.
- sctoolbox.tools.norm_correct.tfidf_normalization(matrix: spmatrix, tf_type: Literal['raw', 'term_frequency', 'log'] = 'term_frequency', idf_type: Literal['unary', 'inverse_freq', 'inverse_freq_smooth'] = 'inverse_freq') csr_matrix[source]
Perform TF-IDF normalization on a sparse matrix.
The different variants of the term frequency and inverse document frequency are obtained from https://en.wikipedia.org/wiki/Tf-idf.
- Parameters:
matrix (scipy.sparse matrix) – The matrix to be normalized.
tf_type (Literal["term_frequency", "log", "raw"], default "term_frequency") – The type of term frequency to use. Can be either “raw”, “term_frequency” or “log”.
idf_type (Literal["inverse_freq", "unary", "inverse_freq_smooth"], default "inverse_freq") – The type of inverse document frequency to use. Can be either “unary”, “inverse_freq” or “inverse_freq_smooth”.
- Returns:
tfidf normalized sparse matrix.
- Return type:
sparse.csr_matrix
Notes
This function requires a lot of memory. Another option is to use the ac.pp.tfidf of the muon package.
- sctoolbox.tools.norm_correct.wrap_corrections(adata: AnnData, batch_key: str, methods: Literal['bbknn', 'combat', 'mnn', 'harmony', 'scanorama'] | list[Literal['bbknn', 'combat', 'mnn', 'harmony', 'scanorama']] | Callable = ['bbknn', 'mnn'], method_kwargs: dict = {}) dict[str, AnnData][source]
Calculate multiple batch corrections for adata using the ‘batch_correction’ function.
- Parameters:
adata (sc.AnnData) – An annotated data matrix object to apply corrections to.
batch_key (str) – The column in adata.obs containing batch information.
methods (list[batch_methods] | Callable | batch_methods, default ["bbknn", "mnn"]) – The method(s) to use for batch correction. Options are: - bbknn - mnn - harmony - scanorama - combat Or provide a custom batch correction function. See batch_correction(method) for more information.
method_kwargs (dict, default {}) – Dict with methods as keys. Values are dicts of additional parameters forwarded to method. See batch_correction(**kwargs).
- Returns:
Dictonary of batch corrected anndata objects. Where the key is the correction method and the value is the corrected anndata.
- Return type:
dict[str, sc.AnnData]
- Raises:
ValueError – If not all methods in methods are valid.
- sctoolbox.tools.norm_correct.batch_correction(adata: AnnData, batch_key: str, method: Literal['bbknn', 'combat', 'mnn', 'harmony', 'scanorama'] | list[Literal['bbknn', 'combat', 'mnn', 'harmony', 'scanorama']] | Callable = ['bbknn', 'mnn'], highly_variable: bool = True, **kwargs: Any) AnnData[source]
Perform batch correction on the adata object using the ‘method’ given.
- Parameters:
adata (sc.AnnData) – An annotated data matrix object to apply corrections to.
batch_key (str) – The column in adata.obs containing batch information.
method (str or function) –
Either one of the predefined methods or a custom function for batch correction. Note: The custom function is expected to accept an anndata object as the first parameter and return the batch corrected anndata.
- Available methods:
bbknn
mnn
harmony
scanorama
combat
highly_variable (bool, default True) – Only for method ‘mnn’. If True, only the highly variable genes (column ‘highly_variable’ in .var) will be used for batch correction.
**kwargs (Any) – Additional arguments will be forwarded to the method function.
- Returns:
A copy of the anndata with applied batch correction.
- Return type:
sc.AnnData
- Raises:
ValueError: –
If batch_key column is not in adata.obs 2. If batch correction method is invalid.
KeyError: – If PCA has not been calculated before running bbknn.
- sctoolbox.tools.norm_correct.evaluate_batch_effect(adata: AnnData, batch_key: str, obsm_key: str = 'X_umap', col_name: str = 'LISI_score', max_dims: int = 5, perplexity: int = 30, inplace: bool = False) AnnData | None[source]
Evaluate batch effect methods using LISI.
- Parameters:
adata (sc.AnnData) – Anndata object with PCA and umap/tsne for batch evaluation.
batch_key (str) – The column in adata.obs containing batch information.
obsm_key (str, default 'X_umap') – The column in adata.obsm containing coordinates.
col_name (str, default 'LISI_score') – Column name for storing the LISI score in .obs.
max_dims (int, default 5) – Maximum number of dimensions of adata.obsm matrix to use for LISI (to speed up computation).
perplexity (int, default 30) – Perplexity for the LISI score calculation.
inplace (bool, default False) – Whether to work inplace on the anndata object.
- Returns:
if inplace is True, LISI_score is added to adata.obs inplace (returns None), otherwise a copy of the adata is returned.
- Return type:
Optional[sc.AnnData]
Notes
LISI score is calculated for each cell and it is between 1-n for a data-frame with n categorical variables.
indicates the effective number of different categories represented in the local neighborhood of each cell.
If the cells are well-mixed, then we expect the LISI score to be near n for a data with n batches.
The higher the LISI score is, the better batch correction method worked to normalize the batch effect and mix the cells from different batches.
For further information on LISI: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1850-9
- Raises:
KeyError: –
If obsm_key is not in adata.obsm. 2. If batch_key is no column in adata.obs.
- sctoolbox.tools.norm_correct.wrap_batch_evaluation(adatas: dict[str, AnnData], batch_key: str, obsm_keys: str | list[str] = ['X_pca', 'X_umap'], threads: int = 1, max_dims: int = 5, inplace: bool = False) dict[str, AnnData] | None[source]
Evaluate batch correction methods for a dict of anndata objects (using LISI score calculation).
- Parameters:
adatas (dict[str, sc.AnnData]) – Dict containing an anndata object for each batch correction method as values. Keys are the name of the respective method. E.g.: {“bbknn”: anndata}
batch_key (str) – The column in adata.obs containing batch information.
obsm_keys (str | list[str], default ['X_pca', 'X_umap']) – Key(s) to coordinates on which the score is calculated.
threads (int, default 1) – Number of threads to use for parallelization.
max_dims (int, default 5) – Maximum number of dimensions of adata.obsm matrix to use for LISI (to speed up computation).
inplace (bool, default False) – Whether to work inplace on the anndata dict.
- Returns:
Dict containing an anndata object for each batch correction method as values of LISI scores added to .obs.
- Return type:
Optional[dict[str, sc.AnnData]]
Analysis
embedding
Embedding tools.
- sctoolbox.tools.embedding.wrap_umap(adatas: Iterable[AnnData], threads: int = 4, **kwargs: Any) None[source]
Compute umap for a list of adatas in parallel.
- Parameters:
adatas (Iterable[sc.AnnData]) – List of anndata objects to compute umap on.
threads (int, default 4) – Number of threads to use.
**kwargs (Any) – Additional arguments to be passed to sc.tl.umap.
- sctoolbox.tools.embedding.correlation_matrix(adata: AnnData, which: Literal['obs', 'var'] = 'obs', basis: str = 'pca', n_components: int | None = None, columns: list[str] | None = None, method: Literal['spearmanr', 'pearsonr'] = 'spearmanr') tuple[DataFrame, DataFrame][source]
Compute a matrix of correlation values between an embedding and given columns.
- Parameters:
adata (sc.AnnData) – Annotated data matrix object.
which (Literal["obs", "var"], default "obs") – Whether to use the observations (“obs”) or variables (“var”) for the correlation.
basis (str, default "pca") – Dimensionality reduction to calculate correlation with. Must be a key in adata.obsm, or a basis available as “X_<basis>” such as “umap”, “tsne” or “pca”.
n_components (int, default None) – Number of components to use for the correlation.
columns (Optional[list[str]], default None) – List of columns to use for the correlation. If None, all numeric columns are used.
method (Literal["spearmanr", "pearson"], default "spearmanr") – Method to use for correlation. Must be either “pearsonr” or “spearmanr”.
- Returns:
correlation coefficient, p-values
- Return type:
Tuple[pd.DataFrame, pd.DataFrame]
- Raises:
ValueError – If “basis” is not found in data, if “which” is not “obs” or “var”, if “method” is not “pearsonr” or “spearmanr”, or if “which” is “var” and “basis” not “pca”.
KeyError – If any of the given columns is not found in the respective table.
clustering
Module for cell clustering.
- sctoolbox.tools.clustering.recluster(adata: AnnData, column: str, clusters: str | list[str], task: Literal['join', 'split'] = 'join', method: Literal['leiden', 'louvain'] = 'leiden', resolution: float | int = 1, key_added: str | None = None, plot: bool = True, embedding: str = 'X_umap') None[source]
Recluster an anndata object based on an existing clustering column in .obs.
- Parameters:
adata (sc.AnnData) – Annotated data matrix.
column (str | list[str]) – Column in adata.obs to use for re-clustering.
clusters (str | list[str]) – Clusters in column to re-cluster.
task (Literal["join", "split"], default "join") – Task to perform. Options are: - “join”: Join clusters in clusters into one cluster. - “split”: Split clusters in clusters are merged and then reclustered using method and resolution.
method (Literal["leiden", "louvain"], default "leiden") – Clustering method to use. Must be one of “leiden” or “louvain”.
resolution (float, default 1) – Resolution parameter for clustering.
key_added (Optional[str], default None) – Name of the new column in adata.obs. If None, the column name is set to <column>_recluster.
plot (bool, default True) – If a plot should be generated of the re-clustering.
embedding (str, default 'X_umap') – Select which embeding should be used.
- Raises:
KeyError: – If the given embeding is not in the data. 1. If the given embedding is not in the data. 2. If given column is not found in adata.obs
ValueError: – If a given cluster is not found in the adata.
Example
import scanpy as sc import sctoolbox adata = sc.datasets.pbmc68k_reduced() sctoolbox.tools.recluster(adata, column="louvain", clusters=["1", "5"], task="join", method="leiden", resolution=1.5, plot=True)
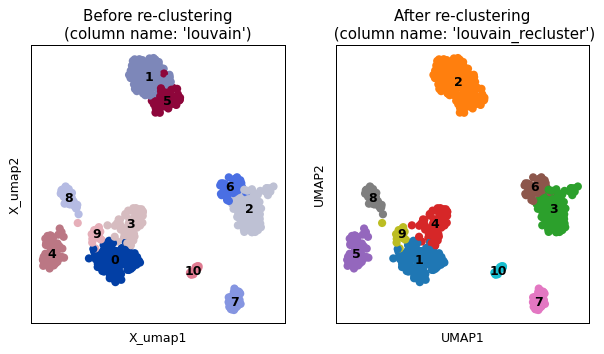
import scanpy as sc import sctoolbox adata = sc.datasets.pbmc68k_reduced() sctoolbox.tools.recluster(adata, column="louvain", clusters=["2", "6"], task="split", method="leiden", resolution=1.5, plot=True)
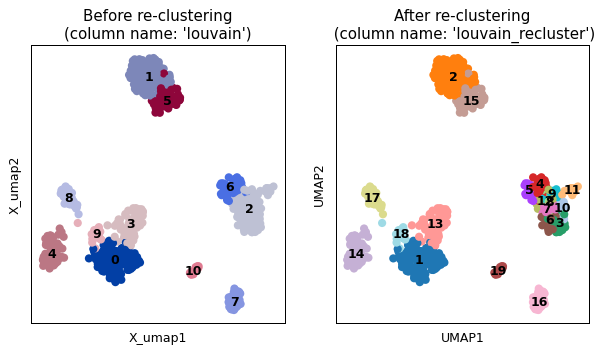
- sctoolbox.tools.clustering.gini(x: _SupportsArray[dtype[Any]] | _NestedSequence[_SupportsArray[dtype[Any]]] | bool | int | float | complex | str | bytes | _NestedSequence[bool | int | float | complex | str | bytes]) float[source]
Calculate the Gini coefficient of a numpy array.
- Parameters:
x (npt.ArrayLike) – Array to calculate Gini coefficient for.
- Returns:
Gini coefficient.
- Return type:
float
- sctoolbox.tools.clustering.calc_ragi(adata: AnnData, condition_column: str = 'clustering', binary_layer: str | None = None) tuple[AnnData, float64][source]
Calculate the RAGI score over all clusters in the adata.
The RAGI score is a measure of how well a cluster is defined by a set of genes. The score is the mean of the Gini coefficients of the gene enrichments across the clusters. The functions uses binary sparse matrices ONLY. If the data is not binary, use sctoolbox.utils.binarize. Binary layers can be selected using the binary_layer parameter. The adata.var table also needs the total counts for each gene.
- Parameters:
adata (sc.AnnData) – Annotated data matrix.
condition_column (str) – Column in adata.obs to use for clustering.
binary_layer (Optional[str], default None) – Layer in adata.layers to use for calculating gene enrichment. If None, the raw layer is used.
- Returns:
Annotated data matrix with the Gini coefficients score in adata.var and RAGI score.
- Return type:
Tuple[sc.AnnData, np.float64]
marker_genes
Tools for marker gene analyis.
- sctoolbox.tools.marker_genes.get_chromosome_genes(gtf: str, chromosomes: str | list[str]) list[str][source]
Get a list of all genes in the gtf for certain chromosome(s).
- Parameters:
gtf (str) – Path to the gtf file.
chromosomes (str | list[str]) – A chromosome or a list of chromosome names to search for genes in.
- Returns:
A list of all genes in the gtf for the given chromosome(s).
- Return type:
list[str]
Notes
This function is not directly used by the framework, but is used to create the marker gene lists for ‘label_genes’.
- Raises:
ValueError: – If not all given chromosomes are found in the GTF-file.
- sctoolbox.tools.marker_genes.label_genes(adata: AnnData, species: str, gene_column: str | None = None) list[str][source]
Label genes as ribosomal, mitochrondrial and gender genes.
Gene labels are added inplace.
- Parameters:
adata (sc.AnnData) – The anndata object.
species (str) – Name of the species.
gene_column (Optional[str], default None) – Name of the column in adata.var that contains the gene names. Uses adata.var.index as default.
- Returns:
List containing the column names added to adata.var.
- Return type:
list[str]
See also
sctoolbox.tools.qc_filter.predict_cell_cyclefor cell cycle prediction.
- sctoolbox.tools.marker_genes.add_gene_expression(adata: AnnData, gene: str) None[source]
Add values of gene/feature per cell to the adata.obs dataframe.
- Parameters:
adata (sc.AnnData) – Anndata object containing gene expression/counts.
gene (str) – Name of the gene/feature from the adata.var index to be added to adata.obs.
- Raises:
ValueError: – If gene is not in adata.var.index.
- sctoolbox.tools.marker_genes.run_rank_genes(adata: AnnData, groupby: str, min_in_group_fraction: float = 0.25, min_fold_change: float = 0.5, max_out_group_fraction: float = 0.8, **kwargs: Any) None[source]
Run scanpy rank_genes_groups and filter_rank_genes_groups.
- Parameters:
adata (sc.AnnData) – Anndata object containing gene expression/counts.
groupby (str) – Column by which the cells in adata should be grouped.
min_in_group_fraction (float, default 0.25) – Minimum fraction of cells in a group that must express a gene to be considered as a marker gene. Parameter forwarded to scanpy.tl.filter_rank_genes_groups.
min_fold_change (float, default 0.5) – Minimum foldchange (+/-) to be considered as a marker gene. Parameter forwarded to scanpy.tl.filter_rank_genes_groups.
max_out_group_fraction (float, default 0.8) – Maximum fraction of cells in other groups that must express a gene to be considered as a marker gene. Parameter forwarded to scanpy.tl.filter_rank_genes_groups.
**kwargs (Any) – Additional arguments forwarded to scanpy.tl.rank_genes_groups.
- Raises:
ValueError: – If number of groups defined by the groupby parameter is < 2.
- sctoolbox.tools.marker_genes.pairwise_rank_genes(adata: AnnData, groupby: str, foldchange_threshold: int | float = 1, min_in_group_fraction: float = 0.25, max_out_group_fraction: float = 0.5, **kwargs: Any) DataFrame[source]
Rank genes pairwise between groups in ‘groupby’.
- Parameters:
adata (sc.AnnData) – Anndata object containing expression data.
groupby (str) – Key in adata.obs containing groups to be compared.
foldchange_threshold (int | float, default 1) – Minimum foldchange (+/-) to be considered as a marker gene.
min_in_group_fraction (float, default 0.25) – Minimum fraction of cells in a group that must express a gene to be considered as a marker gene.
max_out_group_fraction (float, default 0.5) – Maximum fraction of cells in other groups that must express a gene to be considered as a marker gene.
**kwargs (Any) – Additional arguments to be passed to scanpy.tl.rank_genes_groups.
- Returns:
Dataframe containinge the pairwise ranked gened between the groups.
- Return type:
pd.DataFrame
- sctoolbox.tools.marker_genes.get_rank_genes_tables(adata: AnnData, key: str = 'rank_genes_groups', n_genes: int | None = 200, out_group_fractions: bool = False, var_columns: list[str] = [], save_excel: str | None = None) dict[str, DataFrame][source]
Get gene tables containing “rank_genes_groups” genes and information per group (from previously chosen groupby).
- Parameters:
adata (sc.AnnData) – Anndata object containing ranked genes.
key (str, default "rank_genes_groups") – The key in adata.uns to be used for fetching ranked genes.
n_genes (Optional[int], default 200) – Number of genes to be included in the tables. If None, all genes are included.
out_group_fractions (bool, default False) – If True, the output tables will contain additional columns giving the fraction of genes per group.
var_columns (list[str], default []) – List of adata.var columns, which will be added to pandas.DataFrame.
save_excel (Optional[str], default None) – The path to a file for writing the marker gene tables as an excel file (with one sheet per group).
- Returns:
A dictionary with group names as keys, and marker gene tables (pandas DataFrames) per group as values.
- Return type:
dict[str, pd.DataFrame]
- Raises:
ValueError: –
If not all columns given in var_columns are in adata.var. 2. If key cannot be found in adata.uns.
- sctoolbox.tools.marker_genes.mask_rank_genes(adata: AnnData, genes: list[str], key: str = 'rank_genes_groups', inplace: bool = True) AnnData | None[source]
Mask names with “nan” in .uns[key][“names”] if they are found in given ‘genes’.
- Parameters:
adata (sc.AnnData) – Anndata object containing ranked genes.
genes (list[str]) – List of genes to be masked.
key (str, default "rank_genes_groups") – The key in adata.uns to be used for fetching ranked genes.
inplace (bool, default True) – If True, modifies adata.uns[key][“names”] in place. Otherwise, returns a copy of adata.
- Returns:
If inplace = True, modifies adata.uns[key][“names”] in place and returns None. Otherwise, returns a copy of adata.
- Return type:
Optional[sc.AnnData]
- Raises:
ValueError: – If genes is not of type list.
- sctoolbox.tools.marker_genes.run_deseq2(adata: AnnData, sample_col: str, condition_col: str, confounders: list[str] | None = None, layer: str | None = None, percentile_range: tuple[int, int] = (0, 100)) DataFrame[source]
Run DESeq2 on counts within adata. Must be run on the raw counts per sample. If the adata contains normalized counts in .X, ‘layer’ can be used to specify raw counts.
- Parameters:
adata (sc.AnnData) – Anndata object containing raw counts.
sample_col (str) – Column name in adata.obs containing sample names.
condition_col (str) – Column name in adata.obs containing condition names to be compared.
confounders (Optional[list[str]], default None) – List of additional column names in adata.obs containing confounders to be included in the model.
layer (Optional[str], default None) – Name of layer containing raw counts to be used for DESeq2. Default is None (use .X for counts)
percentile_range (Tuple[int, int], default (0, 100)) – Percentile range of cells to be used for calculating pseudobulks. Setting (0,95) will restrict calculation to the cells in the 0-95% percentile ranges. Default is (0, 100), which means all cells are used.
- Returns:
A dataframe containing the results of the DESeq2 analysis. Also adds the dataframe to adata.uns[“deseq_result”]
- Return type:
pd.DataFrame
- Raises:
ValueError: –
If any given column name is not found in adata.obs. 2. If any given column name contains characters not compatible with R.
Notes
Needs the package ‘diffexpr’ to be installed along with ‘bioconductor-deseq2’ and ‘rpy2’. These can be obtained by installing the sctoolbox [deseq2] extra with pip using: pip install . .[deseq2].
See also
sctoolbox.utils.pseudobulk_table
- sctoolbox.tools.marker_genes.score_genes(adata: AnnData, gene_set: str | list[str], score_name: str = 'score', inplace: bool = True, **kwargs: Any) AnnData | None[source]
Assign a score to each cell depending on the expression of a set of genes. This is a wrapper for scanpy.tl.score_genes.
- Parameters:
adata (sc.AnnData) – Anndata object to score.
gene_set (str | list[str]) – A list of genes or path to a file containing a list of genes. The txt file should have one gene per row.
score_name (str, default "score") – Name of the column in obs table where the score will be added.
inplace (bool, default True) – Adds the new column to the original anndata object.
**kwargs (Any) – Additional arguments to be passed to scanpy.tl.score_genes.
- Returns:
If inplace is False, return a copy of anndata object with the new column in the obs table.
- Return type:
Optional[sc.AnnData]
- Raises:
FileNotFoundError: – If path given in gene_set does not lead to a file.
celltype_annotation
Module for general celltype annotation.
- sctoolbox.tools.celltype_annotation.add_cellxgene_annotation(adata: AnnData, csv: str) None[source]
Add columns from cellxgene annotation to the adata .obs table.
- Parameters:
adata (sc.AnnData) – Adata object to add annotations to.
csv (str) – Path to the annotation file from cellxgene containing cell annotation.
- sctoolbox.tools.celltype_annotation.get_celltype_assignment(adata: AnnData, clustering: str, marker_genes_dict: dict[str, list[str]], column_name: str = 'celltype') dict[str, str][source]
Get cell type assignment based on marker genes.
TODO make this more robust
- Parameters:
adata (sc.AnnData) – Anndata object containing raw counts.
clustering (str) – Name of clustering column to use for cell type assignment.
marker_genes_dict (dict[str, list[str]]) – Dictionary containing cell type names as keys and lists of marker genes as values.
column_name (str, default: "celltype") – Name of column to add to adata.obs containing cell type assignment.
- Returns:
Returns a dictionary with cluster-to-celltype mapping (key: cluster name, value: cell type) Also adds the cell type assignment to adata.obs[<column_name>] in place.
- Return type:
dict[str, str]
- sctoolbox.tools.celltype_annotation.run_scsa(adata: AnnData, gene_column: str | None = None, gene_symbol: Literal['auto', 'symbol', 'id'] = 'auto', key: str = 'rank_genes_groups', column_added: str = 'SCSA_pred_celltype', inplace: bool = True, python_path: str | None = None, species: Literal['Human', 'Mouse'] | None = 'Human', fc: float | int = 1.5, pvalue: float = 0.05, tissue: str = 'All', user_db: str | None = None, celltype_column: str = 'cell_name') AnnData | None[source]
Run SCSA cell type annotation and assign cell types to cluster in an adata object.
This is a wrapper function that extracts ranked genes generated by scanpy.tl.rank_genes_groups function and generates input matrix for SCSA, then runs SCSA and assigns cell types to clusters in adata.obs.
Also adds adata.uns[‘SCSA’] as a dictionary with the following keys: - ‘results’: SCSA result table - ‘stderr’: SCSA stderr - ‘stdout’: SCSA stdout - ‘cmd’: SCSA command
Notes
SCSA sometimes gives ValueError: MultiIndex (as covered in https://github.com/bioinfo-ibms-pumc/SCSA/issues/19). This can be solved by downgrading pandas to 1.2.4.
- Parameters:
adata (sc.AnnData) – Adata object to be annotated, must contain ranked genes in adata.uns
gene_column (str, default None) – Name of the column in adata.var that contains the gene names.
gene_symbol (str, default 'auto') – TODO Implement The type of gene symbol. One of “auto”, “symbol” (gene name) or “id” (ensembl id).
key (str, default 'rank_genes_groups') – The key in adata.uns where ranked genes are stored.
column_added (str, default 'SCSA_pred_celltype') – The column name in adata.obs where the cell types will be added.
inplace (bool, default True) – If True, cell types will be added to adata.obs.
python_path (str, default None) – SCSA parameter: Path to python. If not given, will be inferred from sys.executable.
species (Optional[Literal['Human', 'Mouse']], default 'Human') – SCSA parameter: Supports only Human or Mouse. Set to None to use the user defined database given in user_db.
fc (float, default 1.5) – SCSA parameter: Fold change threshold to filter genes.
pvalue (float, default 0.05) – SCSA parameter: P-value threshold to filter genes.
tissue (str, default 'All') – TODO Implement SCSA parameter: A specific tissue can be defined.
user_db (str, default None) – SCSA parameter: Path to the user defined marker database. Must contain at least two columns, one named “cell_name” (or set via celltype_column) for the cell type annotation, and at least one more column with gene names or ids (selected automatically from best gene overlap).
celltype_column (str, default 'cell_name') – SCSA parameter: The column name in the user_db that contains the cell type annotation.
- Returns:
If inplace==False, returns adata with cell types in adata.obs Else return None
- Return type:
Optional[sc.AnnData]
- Raises:
KeyError: –
If key is not in adata.uns. 2. If ‘params’ is not in adata.uns[key] or if ‘groupby’ is not in adata.uns[key][‘params’]. 3. If gene column is not in adata.var
ValueError: –
If species parameter is not Human, Mouse or None. 2. If no species and no user database is provided. 3. If SCSA run failes
Additional
calc_overlap_fc
Module to calculate fold change of reads or fragments from a BAM or fragments file that overlap specified regions.
- sctoolbox.tools.calc_overlap_fc.pct_fragments_in_promoters(adata: AnnData, gtf_file: str | None = None, bam_file: str | None = None, fragments_file: str | None = None, cb_col: str | None = None, cb_tag: str = 'CB', species: Literal['bos_taurus', 'caenorhabditis_elegans', 'canis_lupus_familiaris', 'danio_rerio', 'drosophila_melanogaster', 'gallus_gallus', 'homo_sapiens', 'mus_musculus', 'oryzias_latipes', 'rattus_norvegicus', 'sus_scrofa', 'xenopus_tropicalis'] | None = None, threads: int = 1) None[source]
Calculate the fold change of fragments in promoters.
This function calculates for each cell, the fold change of fragments in a BAM alignment file that overlap with a promoter region specified in a GTF file. The results are added to the anndata object as new column. This is a wrapper function for fc_fragments_in_regions to use promoters.
- Parameters:
adata (sc.AnnData) – The anndata object containig cell barcodes in adata.obs.
gtf_file (str, default None) – Path to GTF file for promoters regions. if None, the GTF file in flatfiles directory will be used.
bam_file (str, default None) – Path to BAM file. If None, a fragments file must be provided in the parameter ‘fragments_file’.
fragments_file (str, default None) – Path to fragments file. If None, a BAM file must be provided in the parameter ‘bam_file’. The BAM file will be converted into fragments file.
cb_col (str, default None) – The column in adata.obs containing cell barcodes. If None, adata.obs.index will be used.
cb_tag (str, default 'CB') – The tag where cell barcodes are saved in the bam file. Set to None if the barcodes are in read names.
species (str, default None) – Name of the species, will only be used if gtf_file is None to use internal GTF files. Species are {bos_taurus, caenorhabditis_elegans, canis_lupus_familiaris, danio_rerio, drosophila_melanogaster, gallus_gallus, homo_sapiens, mus_musculus, oryzias_latipes, rattus_norvegicus, sus_scrofa, xenopus_tropicalis}
threads (int, default 1) – Number of threads for parallelization. Will be used to convert BAM to fragments file.
- Raises:
ValueError – If no species and no gtf_file is given.
Deprecated since version 0.4b: This will be removed in 0.5. Use the overlap_fragments_in_regions function instead.
- sctoolbox.tools.calc_overlap_fc.fc_fragments_in_regions(adata: AnnData, regions_file: str, bam_file: str | None = None, fragments_file: str | None = None, cb_col: str | None = None, cb_tag: str = 'CB', regions_name: str = 'list', threads: int = 4, temp_dir: str | None = None) None[source]
Calculate the fold change of fragments in a region against the background.
This function calculates the fold change of fragments overlapping a region against the background for each cell. The regions are specified in a BED or GTF file and the fragments should be provided by a fragments or BAM file. The results are added to the anndata object as new column.
- Parameters:
adata (sc.AnnData) – The anndata object containig cell barcodes in adata.obs.
regions_file (str) – Path to BED or GTF file containing regions of interest.
bam_file (Optional[str], default None) – Path to BAM file. If None, a fragments file must be provided in the parameter ‘fragments_file’.
fragments_file (Optional[str], default None) – Path to fragments file. If None, a BAM file must be provided in the parameter ‘bam_file’. The BAM file will be converted into fragments file.
cb_col (Optional[str], default None) – The column in adata.obs containing cell barcodes. If None, adata.obs.index will be used.
cb_tag (str, default 'CB') – The tag where cell barcodes are saved in the bam file. Set to None if the barcodes are in read names.
regions_name (str, default 'list') – The name of the regions in the BED or GTF file (e.g. Exons). The name will be used as columns’ name added to the anndata object (e.g. pct_fragments_in_{regions_name}).
threads (int, default 1) – Number of threads for parallelization. Will be used to convert BAM to fragments file.
temp_dir (Optional[str], default None) – Path to temporary directory. Will use the current working directory by default.
- Raises:
ValueError – If bam_file and fragment file is not provided.
- sctoolbox.tools.calc_overlap_fc.count_fragments_per_cell(df: DataFrame, barcode_col: str = 'barcode', frag_count: str = 'count') DataFrame[source]
Get counts per cell from a dataframe containing fragments.
The dataframe must have a column with cell barcodes and a column with fragment counts.
- Parameters:
df (pd.DataFrame) – The dataframe of a bedfile containing the fragments.
barcode_col (str, default 'barcode') – The column name containing the cell barcodes.
frag_count (str, default 'count') – The column name containing the fragment counts.
- Returns:
The dataframe containing the number of fragments per cell.
- Return type:
pd.DataFrame
multiomics
Tools for multiomics analysis.
- sctoolbox.tools.multiomics.merge_anndata(anndata_dict: dict[str, AnnData], join: Literal['inner', 'outer'] = 'inner') AnnData[source]
Merge two h5ad files for dual cellxgene deplyoment.
- Parameters:
anndata_dict (dict[str, sc.AnnData]) – Dictionary with labels as keys and anndata objects as values.
join (Literal['inner', 'outer'], default 'inner') –
Set how to join cells of the adata objects: [‘inner’, ‘outer’]. This only affects the cells since the var/gene section is simply added.
’inner’: only keep overlapping cells.
’outer’: keep all cells. This will add placeholder cells/dots to plots currently disabled.
- Returns:
Merged anndata object.
- Return type:
sc.AnnData
Notes
Important: Depending on the size of the anndata objects the function takes around 60 to 300 GB of RAM! To save RAM and runtime the function generates a minimal anndata object. Only .X, .var, .obs and .obsm are kept. Layers, .varm, etc is removed.
- Raises:
ValueError: – If no indices of both adata.obs tables are overlapping.
bam
Functionality to split bam files into smaller bam files based on clustering in adata.obs.
- sctoolbox.tools.bam.bam_adata_ov(adata: AnnData, bamfile: str, cb_tag: str = 'CB') float[source]
Check if adata.obs barcodes existing in a column of a bamfile.
- Parameters:
adata (sc.AnnData) – adata object where adata.obs is stored
bamfile (str) – path of the bamfile to investigate
cb_tag (str, default 'CB') – bamfile column to extract the barcodes from
- Returns:
hitrate of the barcodes in the bamfile
- Return type:
float
- sctoolbox.tools.bam.check_barcode_tag(adata: AnnData, bamfile: str, cb_tag: str = 'CB') None[source]
Check for the possibilty that the wrong barcode is used.
- Parameters:
adata (sc.AnnData) – adata object where adata.obs is stored
bamfile (str) – path of the bamfile to investigate
cb_tag (str, default 'CB') – bamfile column to extract the barcodes from
- Raises:
ValueError – If barcode hit rate could not be identified
- sctoolbox.tools.bam.subset_bam(bam_in: str, bam_out: str, barcodes: Iterable[str], read_tag: str = 'CB', pysam_threads: int = 4, overwrite: bool = False) None[source]
Subset a bam file based on a list of barcodes.
- Parameters:
bam_in (str) – Path to input bam file.
bam_out (str) – Path to output bam file.
barcodes (Iterable[str]) – List of barcodes to keep.
read_tag (str, default "CB") – Tag in bam file to use for barcode.
pysam_threads (int, default 4) – Number of threads to use for pysam.
overwrite (bool, default False) – Overwrite output file if it exists.
- sctoolbox.tools.bam.split_bam_clusters(adata: AnnData, bams: str | Iterable[str], groupby: str, barcode_col: str | None = None, read_tag: str = 'CB', output_prefix: str = 'split_', reader_threads: int = 1, writer_threads: int = 1, parallel: bool = False, pysam_threads: int = 4, buffer_size: int = 10000, max_queue_size: int = 1000, individual_pbars: bool = False, sort_bams: bool = False, index_bams: bool = False) None[source]
Split BAM files into clusters based on ‘groupby’ from the anndata.obs table.
- Parameters:
adata (sc.Anndata) – Annotated data matrix containing clustered cells in .obs.
bams (str | Iterable[str]) – One or more BAM files to split into clusters
groupby (str) – Name of a column in adata.obs to cluster the pseudobulks by.
barcode_col (str, default None) – Name of a column in adata.obs to use as barcodes. If None, use the index of .obs.
read_tag (str, default "CB") – Tag to use to identify the reads to split. Must match the barcodes of the barcode_col.
output_prefix (str, default split_) – Prefix to use for the output files.
reader_threads (int, default 1) – Number of threads to use for reading.
writer_threads (int, default 1) – Number of threads to use for writing.
parallel (bool, default False) – Whether to enable parallel processsing.
pysam_threads (int, default 4) – Number of threads for pysam.
buffer_size (int, default 10000) – The size of the buffer between readers and writers.
max_queue_size (int, default 1000) – The maximum size of the queue between readers and writers.
individual_pbars (bool, default False) – Whether to show a progress bar for each individual BAM file and output clusters. Default: False (overall progress bars).
sort_bams (bool, default False) – Sort reads in each output bam
index_bams (bool, default False) – Create an index file for each output bam. Will throw an error if sort_bams is False.
- Raises:
ValueError: –
If groupby column is not in adata.obs 2. If barcode column is not in adata.obs 3. If index_bams is set and sort_bams is False
- sctoolbox.tools.bam.open_bam(file: str, mode: str, verbosity: Literal[0, 1, 2, 3] = 3, **kwargs: Any) pysam.AlignmentFile[source]
Open bam file with pysam.AlignmentFile. On a specific verbosity level.
- Parameters:
file (str) – Path to bam file.
mode (str) – Mode to open the file in. See pysam.AlignmentFile
verbosity (Literal[0, 1, 2, 3], default 3) – Set verbosity level. Verbosity level 0 for no messages.
**kwargs (Any) – Forwarded to pysam.AlignmentFile
- Returns:
Object to work on SAM/BAM files.
- Return type:
pysam.AlignmentFile
- sctoolbox.tools.bam.get_bam_reads(bam_obj: pysam.AlignmentFile) int[source]
Get the number of reads from an open pysam.AlignmentFile.
- Parameters:
bam_obj (pysam.AlignmentFile) – An open pysam.AlignmentFile object to get the number of reads from.
- Returns:
number of reads in the bam file
- Return type:
int
- sctoolbox.tools.bam.bam_to_bigwig(bam: str, output: str | None = None, scale: bool = True, overwrite: bool = True, tempdir: str = '.', remove_temp: bool = True, bedtools_path: str | None = None, bgtobw_path: str | None = None) str[source]
Convert reads in a bam-file to bigwig format.
- Parameters:
bam (str) – Path to bam file.
output (Optional[str], default None) – Path to output file. If None, output is written to same directory as bam file with same name and .bw extension.
scale (bool, default True) – Scale output depth to reads per million mapped reads.
overwrite (bool, default True) – Overwrite output file if it already exists.
tempdir (str, default ".") – Path to directory where temporary files are written.
remove_temp (bool, default True) – Remove temporary files after conversion.
bedtools_path (Optional[str], default None) – Path to bedtools binary. If None, the function will search for the binary in the path.
bgtobw_path (Optional[str], default None) – Path to bedGraphToBigWig binary. If None, the function will search for the binary in the path.
- Returns:
Path to output file.
- Return type:
str
- sctoolbox.tools.bam.create_fragment_file(bam: str, barcode_tag: str | None = 'CB', barcode_regex: str | None = None, outdir: str | None = None, nproc: int = 1, index: bool = False, min_dist: int = 10, max_dist: int = 5000, include_clipped: bool = True, shift_plus: int = 5, shift_minus: int = -4, keep_temp: bool = False) str[source]
Create fragments file out of a BAM file.
This is an alternative to using the sinto package, which is slow and has issues with large bam-files.
- Parameters:
bam (str) – Path to .bam file.
barcode_tag (Optional[str], default 'CB') – The tag where cell barcodes are saved in the bam file. Set to None if the barcodes are in read names.
barcode_regex (Optional[str], default None) – Regex to extract barcodes from read names. Set to None if barcodes are stored in a tag.
outdir (Optional[str], default None) – Path to save fragments file. If None, the file will be saved in the same folder as the .bam file. Temporary intermediate files are also written to this directory.
nproc (int, default 1) – Number of threads for parallelization.
index (bool, default False) – If True, index fragments file. Requires bgzip and tabix.
min_dist (int, default 10) – Minimum fragment length to consider.
max_dist (int, default 5000) – Maximum fragment length to consider.
include_clipped (bool, default True) – Whether to include soft-clipped bases in the fragment length. If True, the full length between reads is used. If False, the fragment length will be calculated from the aligned parts of the reads.
shift_plus (int, default 5) – Shift the start position of the forward read by this value (standard for ATAC).
shift_minus (int, default -4) – Shift the end position of the reverse read by this value (standard for ATAC).
keep_temp (bool, default False) – If True, keep temporary files.
- Returns:
Path to fragments file.
- Return type:
str
- Raises:
ValueError – If both barcode_tag and barcode_regex (or neither) are set.
FileNotFoundError – If the input bam file does not exist.
frip
Module containing functions for calculating the FRiP score.
- sctoolbox.tools.frip.calc_frip_scores(adata: AnnData, fragments: str, temp_dir: str = '') tuple[AnnData, float][source]
Calculate the FRiP score for each barcode and adds it to adata.obs.
- Parameters:
adata (sc.AnnData) – AnnData object containing the fragments
fragments (str) – path to fragments bedfile
temp_dir (str, default "") – path to temp directory
- Returns:
AnnData object containing the fragments total FRiP score
- Return type:
Tuple[sc.AnnData, float]
tsse
Module to calculate TSS enrichment scores.
- sctoolbox.tools.tsse.write_TSS_bed(gtf: str, custom_TSS: str, negativ_shift: int = 2000, positiv_shift: int = 2000, temp_dir: str | None = None) tuple[list, list][source]
Write a custom TSS file from a gene gtf file.
negativ_shift and positiv_shift are the number of bases of the flanks. The output is a bed file with the following columns: 1: chr, 2: start, 3:stop and a list of lists with the following columns: 1: chr, 2: start, 3:stop
- Parameters:
gtf (str) – path to gtf file
custom_TSS (str) – path to output file
negativ_shift (int, default 2000) – number of bases to shift upstream
positiv_shift (int, default 2000) – number of bases to shift downstream
temp_dir (Optional[str], default None) – path to temporary directory
- Returns:
list of lists with the following columns: 1: chr, 2: start, 3:stop
- Return type:
Tuple[list, list]
- sctoolbox.tools.tsse.overlap_and_aggregate(fragments: str, custom_TSS: str, overlap: str, tss_list: list[list[str | int]], negativ_shift: int = 2000, positiv_shift: int = 2000) tuple[dict[str, list], list[str]][source]
Overlap the fragments with the custom TSS file and aggregates the fragments in a dictionary.
The dictionary has the following structure: {barcode: [tss_agg, n_fragments]} tss_agg is a numpy array with the aggregated fragments around the TSS with flanks of negativ_shift and positiv_shift. n_fragments is the number of fragments overlapping the TSS.
- Parameters:
fragments (str) – path to fragments file
custom_TSS (str) – path to custom TSS file
overlap (str) – path to output file
tss_list (list[list[str | int]]) – list of lists with the following columns: 1: chr, 2: start, 3:stop
negativ_shift (int, default 2000) – number of bases to shift upstream
positiv_shift (int, default 2000) – number of bases to shift downstream
- Returns:
dictionary with the following structure: {barcode: [tss_agg, n_fragments]} tss_agg is a numpy array with the aggregated fragments around the TSS with flanks of negativ_shift and positiv_shift. n_fragments is the number of fragments overlapping the TSS. list[str] contains a list of temporary files.
- Return type:
Tuple[dict[str, list], list[str]]
- sctoolbox.tools.tsse.calc_per_base_tsse(tSSe_df: DataFrame, min_bias: float = 0.01, edge_size: int = 100) _SupportsArray[dtype[Any]] | _NestedSequence[_SupportsArray[dtype[Any]]] | bool | int | float | complex | str | bytes | _NestedSequence[bool | int | float | complex | str | bytes][source]
Calculate per base tSSe by dividing the tSSe by the bias. The bias is calculated by averaging the edges of the tSSe.
The edges are defined by the edge_size.
- Parameters:
tSSe_df (pd.DataFrame) – dataframe with the following columns: 1: barcode, 2: tSSe, 3: n_fragments
min_bias (float, default 0.01) – minimum bias to avoid division by zero
edge_size (int, default 100) – number of bases to use for the edges
- Returns:
numpy array with the per base tSSe
- Return type:
npt.ArrayLike
- sctoolbox.tools.tsse.global_tsse_score(per_base_tsse: _SupportsArray[dtype[Any]] | _NestedSequence[_SupportsArray[dtype[Any]]] | bool | int | float | complex | str | bytes | _NestedSequence[bool | int | float | complex | str | bytes], negativ_shift: int, edge_size: int = 50) _SupportsArray[dtype[Any]] | _NestedSequence[_SupportsArray[dtype[Any]]] | bool | int | float | complex | str | bytes | _NestedSequence[bool | int | float | complex | str | bytes][source]
Calculate the global tSSe score by averaging the per base tSSe around the TSS.
- Parameters:
per_base_tsse (npt.ArrayLike) – numpy array with the per base tSSe
negativ_shift (int) – number of bases to shift upstream
edge_size (int, default 50) – number of bases to use for the edges
- Returns:
numpy array with the global tSSe score
- Return type:
npt.ArrayLike
- sctoolbox.tools.tsse.tsse_scoring(fragments: str, gtf: str, negativ_shift: int = 2000, positiv_shift: int = 2000, edge_size_total: int = 100, edge_size_per_base: int = 50, min_bias: float = 0.01, keep_tmp: bool = False, temp_dir: str = '') DataFrame[source]
Calculate the tSSe score for each cell.
Calculating the TSSe score is done like described in: “Chromatin accessibility profiling by ATAC-seq” Fiorella et al. 2022
- Parameters:
fragments (str) – path to fragments file
gtf (str) – path to gtf file
negativ_shift (int, default 2000) – number of bases to shift upstream
positiv_shift (int, default 2000) – number of bases to shift downstream
edge_size_total (int, default 100) – number of bases to use for the edges for the global tSSe score
edge_size_per_base (int, default 50) – number of bases to use for the edges for the per base tSSe score
min_bias (float, default 0.01) – minimum bias to avoid division by zero
keep_tmp (bool, default False) – keep temporary files
temp_dir (str, default "") – path to temporary directory
- Returns:
dataframe with the following columns: 1: barcode, 2: tSSe, 3: n_fragments
- Return type:
pd.DataFrame
- sctoolbox.tools.tsse.add_tsse_score(adata: AnnData, fragments: str, gtf: str, negativ_shift: int = 2000, positiv_shift: int = 2000, edge_size_total: int = 100, edge_size_per_base: int = 50, min_bias: float = 0.01, keep_tmp: bool = False, temp_dir: str = '') AnnData[source]
Add the tSSe score to the adata object.
Calculating the TSSe score is done like described in: “Chromatin accessibility profiling by ATAC-seq” Fiorella et al. 2022
- Parameters:
adata (sc.AnnData) – AnnData object
fragments (str) – path to fragments.bed
gtf (str) – path to gtf file
negativ_shift (int, default 2000) – number of bases to shift upstream for the flanking regions
positiv_shift (int, default 2000) – number of bases to shift downstream for the flanking regions
edge_size_total (int, default 100) – number of bases to use for the edges for the global tSSe score
edge_size_per_base (int, default 50) – number of bases to use for the edges for the per base tSSe score
min_bias (float, default 0.01) – minimum bias to avoid division by zero
keep_tmp (bool, default False) – keep temporary files
temp_dir (str, default "") – path to temporary directory
- Returns:
AnnData object with added tSSe score
- Return type:
sc.AnnData
insertsize
Tools to calculate fragemnt- and insertsize for scATAC.
- sctoolbox.tools.insertsize.add_insertsize(adata: AnnData, bam: str | None = None, fragments: str | None = None, barcode_col: str | None = None, barcode_tag: str = 'CB', regions: str | None = None) None[source]
Add information on insertsize to the adata object using either a .bam-file or a fragments file.
Adds columns “insertsize_count” and “mean_insertsize” to adata.obs and a key “insertsize_distribution” to adata.uns containing the insertsize distribution as a pandas dataframe.
- Parameters:
adata (sc.AnnData) – AnnData object to add insertsize information to.
bam (Optional[str], default None) – Path to bam file containing paired-end reads. If None, the fragments file is used instead.
fragments (Optional[str], default None) – Path to fragments file containing fragments information. If None, the bam file is used instead.
barcode_col (Optional[str], default None) – Column in adata.obs containing the name of the cell barcode. If barcode_col is None, it is assumed that the index of adata.obs contains the barcode.
barcode_tag (str, default 'CB') – Only for bamfiles: Tag used for the cell barcode for each read.
regions (Optional[str], default None) – Only for bamfiles: A list of regions to obtain reads from, e.g. [‘chr1:1-2000000’]. If None, all reads in the .bam-file are used.
- Raises:
ValueError: –
If bam and fragments is given. 2. If bam and fragments is not given. 3. If no barcodes between bam- or fragment-file and adata overlap
peak_annotation
Tools for peak annotation required by scATAC-seq.
- sctoolbox.tools.peak_annotation.annotate_adata(adata: AnnData, gtf: str, config: dict[str, Any] | None = None, best: bool = True, threads: int = 1, coordinate_cols: list[str] | None = None, temp_dir: str = '', remove_temp: bool = True, inplace: bool = True) AnnData | None[source]
Annotate adata .var features with genes from .gtf using UROPA [1].
The function assumes that the adata.var contains genomic coordinates in the first three columns, e.g. “chromosome”, “start”, “end”. If specific columns should be used, please adjust the names via ‘coordinate_cols’.
- Parameters:
adata (sc.AnnData) – The anndata object containing features to annotate.
gtf (str) – Path to .gtf file containing genomic elements for annotation.
config (Optional[dict[str, Any]], default None) – A dictionary indicating how regions should be annotated. Default (None) is to annotate feature ‘gene’ within -10000;1000bp of the gene start. See ‘Examples’ of how to set up a custom configuration dictionary.
best (bool, default True) – Whether to return the best annotation or all valid annotations.
threads (int, default 1) – Number of threads to use for multiprocessing.
coordinate_cols (Optional[list[str]], default None) – A list of column names in the regions DataFrame that contain the chromosome, start and end coordinates. If None the first three columns are taken.
temp_dir (str, default '') – Path to a directory to store files. Is only used if input .gtf-file needs sorting.
remove_temp (boolean, default True) – If True remove temporary directory after execution.
inplace (boolean, default True) – Whether to add the annotations to the adata object in place.
- Returns:
If inplace == True, the annotation is added to adata.var in place. Else, a copy of the adata object is returned with the annotations added.
- Return type:
Optional[sc.AnnData]
References
Examples
>>> custom_config = {"queries": [{"distance": [10000, 1000], "feature_anchor": "start", "feature": "gene"}], "priority": True, "show_attributes": "all"}
>>> annotate_regions(adata, gtf="genes.gtf", config=custom_config)
- sctoolbox.tools.peak_annotation.annotate_narrowPeak(filepath: str, gtf: str, config: dict[str, Any] | None = None, best: bool = True, threads: int = 1, temp_dir: str = '', remove_temp: bool = True) DataFrame[source]
Annotate narrowPeak files with genes from .gtf using UROPA.
- Parameters:
filepath (str) – Path to the narrowPeak file to be annotated.
gtf (str) – Path to the .gtf file containing the genes to be annotated.
config (Optional[dict[str, Any]], default None) – A dictionary indicating how regions should be annotated. Default (None) is to annotate feature ‘gene’ within -10000;1000bp of the gene start. See ‘Examples’ of how to set up a custom configuration dictionary.
best (bool, default True) – Whether to return the best annotation or all valid annotations.
threads (int, default 1) – Number of threads to perform the annotation.
temp_dir (str, default '') – Path to the directory where the temporary files should be written.
remove_temp (bool, default True) – If True remove temporary directory after execution.
- Returns:
Dataframe containing the annotations.
- Return type:
pd.DataFrame
receptor_ligand
Tools for a receptor-ligand analysis.
- sctoolbox.tools.receptor_ligand.download_db(adata: AnnData, db_path: str, ligand_column: str, receptor_column: str, sep: str = '\t', inplace: bool = False, overwrite: bool = False) AnnData | None[source]
Download table of receptor-ligand interactions and store in adata.
- Parameters:
adata (sc.AnnData) – Analysis object the database will be added to.
db_path (str) – Path to database table. A valid database needs a column with receptor gene ids/symbols and ligand gene ids/symbols. Human: http://tcm.zju.edu.cn/celltalkdb/download/processed_data/human_lr_pair.txt Mouse: http://tcm.zju.edu.cn/celltalkdb/download/processed_data/mouse_lr_pair.txt
ligand_column (str) – Name of the column with ligand gene names. Use ‘ligand_gene_symbol’ for the urls provided above.
receptor_column (str) – Name of column with receptor gene names. Use ‘receptor_gene_symbol’ for the urls provided above.
sep (str, default 't') – Separator of database table.
inplace (bool, default False) – Whether to copy adata or modify it inplace.
overwrite (bool, default False) – If True will overwrite existing database.
Notes
This will remove all information stored in adata.uns[‘receptor-ligand’]
- Returns:
If not inplace, return copy of adata with added database path and database table to adata.uns[‘receptor-ligand’]
- Return type:
Optional[sc.AnnData]
- Raises:
ValueError: – 1: If ligand_column is not in database. 2: If receptor_column is not in database.
- sctoolbox.tools.receptor_ligand.calculate_interaction_table(adata: AnnData, cluster_column: str, gene_index: str | None = None, normalize: int = 1000, inplace: bool = False, overwrite: bool = False) AnnData | None[source]
Calculate an interaction table of the clusters defined in adata.
- Parameters:
adata (sc.AnnData) – AnnData object that holds the expression values and clustering
cluster_column (str) – Name of the cluster column in adata.obs.
gene_index (Optional[str], default None) – Column in adata.var that holds gene symbols/ ids. Corresponds to download_db(ligand_column, receptor_column). Uses index when None.
normalize (int, default 1000) – Correct clusters to given size.
inplace (bool, default False) – Whether to copy adata or modify it inplace.
overwrite (bool, default False) – If True will overwrite existing interaction table.
- Returns:
If not inpalce, return copy of adata with added interactions table to adata.uns[‘receptor-ligand’][‘interactions’]
- Return type:
Optional[sc.AnnData]
- Raises:
ValueError: – 1: If receptor-ligand database cannot be found. 2: Id database genes do not match adata genes.
Exception: – If not interactions were found.
- sctoolbox.tools.receptor_ligand.interaction_violin_plot(adata: AnnData, min_perc: int | float, save: str | None = None, figsize: tuple[int, int] = (5, 20), dpi: int = 100) _SupportsArray[dtype[Any]] | _NestedSequence[_SupportsArray[dtype[Any]]] | bool | int | float | complex | str | bytes | _NestedSequence[bool | int | float | complex | str | bytes][source]
Generate violin plot of pairwise cluster interactions.
- Parameters:
adata (sc.AnnData) – AnnData object
min_perc (int | float) – Minimum percentage of cells in a cluster that express the respective gene. A value from 0-100.
save (str, default None) – Output filename. Uses the internal ‘sctoolbox.settings.figure_dir’.
figsize (int tuple, default (5, 20)) – Figure size
dpi (float, default 100) – The resolution of the figure in dots-per-inch.
- Returns:
Object containing all plots. As returned by matplotlib.pyplot.subplots
- Return type:
npt.ArrayLike
- sctoolbox.tools.receptor_ligand.hairball(adata: AnnData, min_perc: int | float, interaction_score: float | int = 0, interaction_perc: int | float | None = None, save: str | None = None, title: str | None = 'Network', color_min: float | int = 0, color_max: int | float | None = None, cbar_label: str = 'Interaction count', show_count: bool = False, restrict_to: list[str] | None = None, additional_nodes: list[str] | None = None, hide_edges: list[tuple[str, str]] | None = None) _SupportsArray[dtype[Any]] | _NestedSequence[_SupportsArray[dtype[Any]]] | bool | int | float | complex | str | bytes | _NestedSequence[bool | int | float | complex | str | bytes][source]
Generate network graph of interactions between clusters.
- Parameters:
adata (sc.AnnData) – AnnData object
min_perc (int | float) – Minimum percentage of cells in a cluster that express the respective gene. A value from 0-100.
interaction_score (float | int, default 0) – Interaction score must be above this threshold for the interaction to be counted in the graph.
interaction_perc (Optional[int | float], default None) – Select interaction scores above or equal to the given percentile. Will overwrite parameter interaction_score. A value from 0-100.
save (str, default None) – Output filename. Uses the internal ‘sctoolbox.settings.figure_dir’.
title (str, default 'Network') – The plots title.
color_min (float, default 0) – Min value for color range.
color_max (Optional[float | int], default None) – Max value for color range.
cbar_label (str, default 'Interaction count') – Label above the colorbar.
show_count (bool, default False) – Show the interaction count in the hairball.
restrict_to (Optional[list[str]], default None) – Only show given clusters provided in list.
additional_nodes (Optional[list[str]], default None) – List of additional node names displayed in the hairball.
hide_edges (Optional[list[Tuple[str, str]]], default None) – List of tuples with node names that should not have an edge shown. Order doesn’t matter. E.g. [(“a”, “b”)] to omit the edge between node a and b.
- Returns:
Object containing all plots. As returned by matplotlib.pyplot.subplots
- Return type:
npt.ArrayLike
- Raises:
ValueError: – If restrict_to contains invalid clusters.
- sctoolbox.tools.receptor_ligand.progress_violins(datalist: list[DataFrame], datalabel: list[str], cluster_a: str, cluster_b: str, min_perc: float | int, save: str, figsize: tuple[int | float, int | float] = (12, 6)) str[source]
Show cluster interactions over timepoints.
CURRENTLY NOT FUNCTIONAL!
TODO Implement function
- Parameters:
datalist (list[pd.DataFrame]) – List of interaction DataFrames. Each DataFrame represents a timepoint.
datalabel (list[str]) – List of strings. Used to label the violins.
cluster_a (str) – Name of the first interacting cluster.
cluster_b (str) – Name of the second interacting cluster.
min_perc (float | int) – Minimum percentage of cells in a cluster each gene must be expressed in.
save (str) – Path to output file.
figsize (Tuple[int, int], default (12, 6)) – Tuple of plot (width, height).
- Return type:
str
- sctoolbox.tools.receptor_ligand.interaction_progress(datalist: list[AnnData], datalabel: list[str], receptor: str, ligand: str, receptor_cluster: str, ligand_cluster: str, figsize: tuple[int | float, int | float] = (4, 4), dpi: int = 100, save: str | None = None) Axes[source]
Barplot that shows the interaction score of a single interaction between two given clusters over multiple datasets.
TODO add checks & error messages
- Parameters:
datalist (list[sc.AnnData]) – List of anndata objects.
datalabel (list[str]) – List of labels for the given datalist.
receptor (str) – Name of the receptor gene.
ligand (str) – Name of the ligand gene.
receptor_cluster (str) – Name of the receptor cluster.
ligand_cluster (str) – Name of the ligand cluster.
figsize (Tuple[int | float, int | float], default (4, 4)) – Figure size in inch.
dpi (int, default 100) – Dots per inch.
save (Optional[str], default None) – Output filename. Uses the internal ‘sctoolbox.settings.figure_dir’.
- Returns:
The plotting object.
- Return type:
matplotlib.axes.Axes
- sctoolbox.tools.receptor_ligand.connectionPlot(adata: AnnData, restrict_to: list[str] | None = None, figsize: tuple[int | float, int | float] = (10, 15), dpi: int = 100, connection_alpha: str | None = 'interaction_score', save: str | None = None, title: str | None = None, receptor_cluster_col: str = 'receptor_cluster', receptor_col: str = 'receptor_gene', receptor_hue: str = 'receptor_score', receptor_size: str = 'receptor_percent', receptor_genes: list[str] | None = None, ligand_cluster_col: str = 'ligand_cluster', ligand_col: str = 'ligand_gene', ligand_hue: str = 'ligand_score', ligand_size: str = 'ligand_percent', ligand_genes: list[str] | None = None, filter: str | None = None, lw_multiplier: int | float = 2, wspace: float = 0.4, line_colors: str | None = 'rainbow') _SupportsArray[dtype[Any]] | _NestedSequence[_SupportsArray[dtype[Any]]] | bool | int | float | complex | str | bytes | _NestedSequence[bool | int | float | complex | str | bytes][source]
Show specific receptor-ligand connections between clusters.
- Parameters:
adata (sc.AnnData) – AnnData object
restrict_to (Optional[list[str]], default None) – Restrict plot to given cluster names.
figsize (Tuple[int | float, int | float], default (10, 15)) – Figure size
dpi (float, default 100) – The resolution of the figure in dots-per-inch.
connection_alpha (str, default 'interaction_score') – Name of column that sets alpha value of lines between plots. None to disable.
save (Optional[str], default None) – Output filename. Uses the internal ‘sctoolbox.settings.figure_dir’.
title (Optional[str], default None) – Title of the plot
receptor_cluster_col (str, default 'receptor_cluster') – Name of column containing cluster names of receptors. Shown on x-axis.
receptor_col (str, default 'receptor_gene') – Name of column containing gene names of receptors. Shown on y-axis.
receptor_hue (str, default 'receptor_score') – Name of column containing receptor scores. Shown as point color.
receptor_size (str, default 'receptor_percent') – Name of column containing receptor expression percentage. Shown as point size.
receptor_genes (Optional[list[str]], default None) – Restrict receptors to given genes.
ligand_cluster_col (str, default 'ligand_cluster') – Name of column containing cluster names of ligands. Shown on x-axis.
ligand_col (str, default 'ligand_gene') – Name of column containing gene names of ligands. Shown on y-axis.
ligand_hue (str, default 'ligand_score') – Name of column containing ligand scores. Shown as point color.
ligand_size (str, default 'ligand_percent') – Name of column containing ligand expression percentage. Shown as point size.
ligand_genes (Optional[list[str]], default None) – Restrict ligands to given genes.
filter (Optional[str], default None) – Conditions to filter the interaction table on. E.g. ‘column_name > 5 & other_column < 2’. Forwarded to pandas.DataFrame.query.
lw_multiplier (int | float, default 2) – Linewidth multiplier.
wspace (float, default 0.4) – Width between plots. Fraction of total width.
line_colors (Optional[str], default 'rainbow') – Name of colormap used to color lines. All lines are black if None.
- Returns:
Object containing all plots. As returned by matplotlib.pyplot.subplots
- Return type:
npt.ArrayLike
- Raises:
Exception: – If no onteractions between clsuters are found.
- sctoolbox.tools.receptor_ligand.get_interactions(anndata: AnnData, min_perc: int | float | None = None, interaction_score: int | float | None = None, interaction_perc: int | float | None = None, group_a: list[str] | None = None, group_b: list[str] | None = None, save: str | None = None) DataFrame[source]
Get interaction table from anndata and apply filters.
- Parameters:
anndata (sc.AnnData) – Anndata object to pull interaction table from.
min_perc (Optional[float | int], default None) – Minimum percent of cells in a cluster that express the ligand/ receptor gene. Value from 0-100.
interaction_score (Optional[float | int], default None) – Filter receptor-ligand interactions below given score. Ignored if interaction_perc is set.
interaction_perc (Optional[float | int], default None) – Filter receptor-ligand interactions below the given percentile. Overwrite interaction_score. Value from 0-100.
group_a (Optional[list[str]], default None) – List of cluster names that must be present in any given receptor-ligand interaction.
group_b (Optional[list[str]], default None) – List of cluster names that must be present in any given receptor-ligand interaction.
save (Optional[str], default None) – Output filename. Uses the internal ‘sctoolbox.settings.table_dir’.
- Returns:
Table that contains interactions. Columns:
receptor_cluster = name of the receptor cluster
ligand_cluster = name of the ligand cluster
receptor_gene = name of the receptor gene
ligand_gene = name of the ligand gene
receptor_score = zscore of receptor gene cluster mean expression (scaled by cluster size)
ligand_score = zscore of ligand gene cluster mean expression (scaled by cluster size)
receptor_percent = percent of cells in cluster expressing receptor gene
ligand_percent = percent of cells in cluster expressing ligand gene
receptor_cluster_size = number of cells in receptor cluster
ligand_cluster_size = number of cells in ligand cluster
interaction_score = sum of receptor_score and ligand_score
- Return type:
pd.DataFrame
gene_correlation
Tools for gene-gene correlation.
- sctoolbox.tools.gene_correlation.correlate_conditions(adata: AnnData, gene: str, condition_col: str, condition_A: str, condition_B: str) DataFrame[source]
Calculate the correlation of a gene expression over two conditions and compares the two conditions.
- Parameters:
adata (sc.AnnData) – Annotated adata object.
gene (str) – Gene of interest.
condition_col (str) – Column in adata.obs containing conditions.
condition_A (str) – Name of the first condition.
condition_B (str) – Name of the second condition.
- Returns:
Dataframe containing the correlation of a gene expression over two conditions.
- Return type:
pd.DataFrame
- Raises:
ValueError – If one or both condition columns are not in adata.obs.
- sctoolbox.tools.gene_correlation.correlate_ref_vs_all(adata: AnnData, ref_gene: str, correlation_threshold: float = 0.4, save: str | None = None) DataFrame[source]
Calculate the correlation of the reference gene vs all other genes.
Additionally, plots umap highlighting correlating gene expression.
- Parameters:
adata (sc.AnnData) – Annotated data matrix.
ref_gene (str) – Reference gene.
correlation_threshold (float, default 0.4) – Threshold for correlation value. Correlating genes with a correlation >= threshold will be plotted if plot parameter is set to True.
save (str, default None) – Path to save the figure to.
- Returns:
Dataframe containing correlation of refrence gene to other genes.
- Return type:
pd.DataFrame
- sctoolbox.tools.gene_correlation.compare_two_conditons(df_cond_A: DataFrame, df_cond_B: DataFrame, n_cells_A: int, n_cells_B: int) DataFrame[source]
Compare two conditions.
- Parameters:
df_cond_A (pd.DataFrame) – Dataframe containing correlation analysis from correlate_ref_vs_all
df_cond_B (pd.DataFrame) – Dataframe containing correlation analysis from correlate_ref_vs_all
n_cells_A (int) – Number of cells within condition A
n_cells_B (int) – Number of cells within condition B
- Returns:
Dataframe containing single correlation and Fischer Z transformation
- Return type:
pd.DataFrame
tobias
Tools for TOBIAS usage.
- sctoolbox.tools.tobias.write_TOBIAS_config(out_path: str, bams: list[str] = [], names: list[str] | None = None, fasta: str | None = None, blacklist: str | None = None, gtf: str | None = None, motifs: str | None = None, organism: Literal['human', 'mouse', 'zebrafish'] = 'human', output: str = 'TOBIAS_output', plot_comparison: bool = True, plot_correction: bool = True, plot_venn: bool = True, coverage: bool = True, wilson: bool = True) None[source]
Write a TOBIAS config file from input bams/fasta/blacklist etc.
- Parameters:
out_path (str) – Path to output yaml file.
bams (list[str], default []) – List of paths to bam files.
names (Optional[list[str]], default None) – List of names for the bams. If None, the names are set to the bam file names with common prefix and suffix removed.
fasta (Optional[str], default None) – Path to fasta file.
blacklist (Optional[str], default None) – Path to blacklist file.
gtf (Optional[str], default None) – Path to gtf file.
motifs (Optional[str], default None) – Path to motif file.
organism (Literal["human", "mouse", "zebrafish"], default 'human') – Organism name. TOBIAS supports ‘human’, ‘mouse’ or ‘zebrafish’.
output (str, default 'Tobias_output') – Output directory of the TOBIAS run.
plot_comparison (bool, default True) – Flag for the step of plotting comparison of the TOBIAS run.
plot_correction (bool, default True) – Flag for the step of plotting correction of the TOBIAS run.
plot_venn (bool, default True) – Flag for the step of plotting venn diagramms of the TOBIAS run.
coverage (bool, default True) – Flag for coverage step of the TOBIAS run.
wilson (bool, default True) – Flag for wilson step of the TOBIAS run.